La sECJ sigue siendo una enfermedad rara, ya que afecta a aproximadamente 1 de cada 1 millón de casos al año en todo el mundo.

Una mujer de 56 años, con antecedentes de hipertensión, comenzó a experimentar dolores de cabeza y episodios de desorientación.
En su primera evaluación médica, los análisis de sangre y un TAC de cerebro no mostraron anormalidades, por lo que fue tratada inicialmente por cefalea tensional. Sin embargo, en los dos meses siguientes, su condición empeoró.
Desarrolló dificultad visual, desorientación constante y un rápido deterioro de sus capacidades cognitivas.
Los análisis del líquido cefalorraquídeo (LCR) mostraron resultados normales, pero las proteínas 14-3-3 y tau fueron positivas, indicadores de daño cerebral.
Cuando ingresó en una institución especializada, su estado había empeorado considerablemente: apenas respondía a estímulos dolorosos, no podía comer y sufría neumonía por aspiración y deshidratación, lo que también le causó una insuficiencia renal aguda.
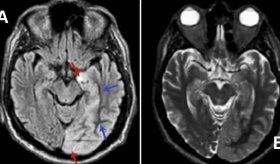
Los estudios adicionales incluyeron una resonancia magnética cerebral, que mostró un patrón típico de la enfermedad de Creutzfeldt-Jakob (ECJ), conocido como el "signo de la cinta cortical".
Además, un electroencefalograma (EEG) reveló actividad cerebral característica de esta enfermedad, con ondas agudas periódicas de 1 Hz. La familia fue informada sobre el pronóstico mortal de la enfermedad y decidió emitir una orden de no reanimar/no intubar. La paciente falleció dos meses después de la aparición de los síntomas.
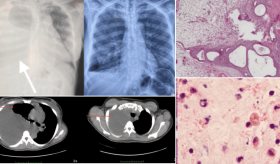
Tras el fallecimiento, se realizó una autopsia con el apoyo del National Prion Disease Pathology Surveillance Center (NPDPSC) en Ohio, ya que no había experiencia local para este procedimiento en Puerto Rico.
El análisis genético y la histopatología confirmaron el diagnóstico de ECJ esporádica. Los hallazgos incluyeron daños cerebrales típicos de esta enfermedad: degeneración espongiforme, gliosis e inmunotinción positiva para la proteína priónica (PrP).
La ECJ esporádica es una enfermedad rara, invariablemente mortal, que afecta al sistema nervioso central. No se identificaron factores de riesgo claros en esta paciente, más allá de una transfusión de sangre recibida muchos años antes.
Actualmente, no existe tratamiento para esta enfermedad, y su manejo se enfoca en aliviar los síntomas y brindar soporte al paciente y a su familia.
El diagnóstico de la enfermedad de Creutzfeldt-Jakob (ECJ) suele ser un reto para la mayoría de los médicos dada su bajísima incidencia y sus diferentes presentaciones clínico-patológicas.
Las enfermedades priónicas son trastornos neurodegenerativos transmisibles con diferentes etiologías ya que pueden ser esporádicas, genéticas o adquiridas por infección.
La enfermedad de Creutzfeldt-Jakob esporádica (ECJes) es la forma más común de las enfermedades priónicas humanas esporádicas. A pesar de ello, la sECJ sigue siendo una enfermedad rara, ya que afecta a aproximadamente 1 de cada 1 millón de casos al año en todo el mundo.
En la sECJ y otras enfermedades priónicas humanas, el codón 129 del gen de la proteína priónica (PrP), el lugar de un polimorfismo común metionina (M)/valina (V), y los dos tipos diferentes de la proteína priónica del prurigo lumbar (PrPSc), se consideran modificadores del fenotipo de la enfermedad.
Las combinaciones del genotipo del codón 129 (129MM, 129MV y 129VV) y el tipo de PrPSc (tipo 1 o tipo 2) se han utilizado para clasificar la sCJD en cinco subtipos diferentes. La sCJDMM1 (por ejemplo, 129MM y PrPSc tipo 1) es el subtipo más común y representa el 55-70% de todas las enfermedades priónicas esporádicas.
Tras la revisión de la literatura, el presente caso ( Del Pilar-Morales et al), representa el primer informe detallado de un paciente con sECJ nativo de Puerto Rico.
Las características clínicas e histopatológicas de este caso encajan bien con las del subtipo sCJDMM1, según confirman los datos genéticos y moleculares llevados a cabo por el NPDPSC. En el banco de datos del NPDPSC se recogieron otros dos casos de sECJ de pacientes nativos y residentes en Puerto Rico.
Estos dos casos tenían el genotipo 129MM (edad de inicio de la enfermedad: 63 años; duración de la enfermedad: 8 meses) o 129MV (edad de inicio de la enfermedad: 66 años; duración de la enfermedad: 8 meses) y ambos eran portadores de PrPres tipo 1.
Así pues, los tres casos de ECJs nativos de Puerto Rico pertenecen al subtipo sCJDMM(MV)1.
Se han propuesto varios criterios diagnósticos para un diagnóstico «probable» de sECJ (5-7). El Centro para el Control y la Prevención de Enfermedades (CDC) describe todos los criterios para la sECJ probable.
Existía la preocupación de que el paciente pudiera haber desarrollado la sECJ debido a una transfusión de sangre recibida varios años antes de la aparición de la enfermedad. Sin embargo, los estudios epidemiológicos, aunque limitados, no han aportado hasta la fecha pruebas de transmisión de la sECJ por transfusión sanguínea (8-9).
Así pues, en este paciente nativo de Puerto Rico, el riesgo potencial de haber adquirido la enfermedad por transfusión sanguínea es improbable. Este informe representa la primera descripción detallada de la sECJ en una familia puertorriqueña.