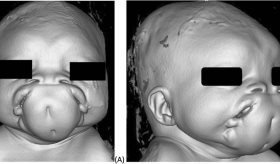
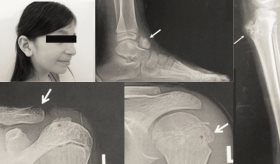
El paciente, con características físicas como braquidactilia y baja estatura, sufría de huesos débiles e infecciones respiratorias recurrentes. Además de hipocalcemia, hiperfosfatemia y niveles elevados de PTH.

Joven de 18 años, acudió a consulta externa de endocrinología por un cuadro clínico complejo que incluía episodios de convulsiones y trastornos metabólicos, particularmente de fosfato y calcio.
Estos síntomas se habían evidenciado durante una hospitalización previa de dos meses en la que se le había diagnosticado hipocalcemia (baja de calcio en sangre), hiperfosfatemia ( exceso de fósforo) y niveles elevados de la hormona paratiroidea (PTH).
El paciente es hijo de padres no consanguíneos, sanos y sin antecedentes familiares significativos de enfermedades endocrinas o metabólicas. Sin embargo, nació prematuro a las 32 semanas de gestación, con un peso al nacer de 2700 g y una talla de 49 cm.
Su desarrollo inicial fue complejo, pues presentó dificultades para la succión y la lactancia materna en los primeros días de vida, lo cual se resolvió con el tiempo. A pesar de superar esta fase, la madre observó un aumento rápido de peso en la infancia, asociado con obesidad infantil, que fue un signo temprano de problemas metabólicos.
A lo largo de su infancia, el paciente desarrolló características físicas notables, como la presencia de braquidactilia (dedos cortos) y una baja estatura, que generaron preocupación en la familia.
A los 9 años, sufrió una fractura patológica del radio izquierdo sin trauma evidente, lo que sugirió fragilidad ósea subyacente. Además, a esa edad comenzaron a manifestarse otras complicaciones, incluyendo infecciones recurrentes del tracto respiratorio superior, que requirieron hospitalización.
A los 14 años, el paciente presentó un episodio clínico más grave, en el que se documentó hipocalcemia (bajos niveles de calcio en sangre), hiperfosfatemia (niveles elevados de fosfato) y niveles elevados de PTH.
Estos hallazgos indicaban disfunción en la regulación del calcio, lo que sugirió la posibilidad de un trastorno endocrino. En ese momento, el paciente fue ingresado en el servicio de urgencias debido a síntomas como parestesias en las extremidades, estreñimiento crónico, epistaxis recurrente y dolor lumbar.
Además, los resultados de las pruebas bioquímicas de sangre mostraron niveles elevados de PTH (409,7 pg/mL, valor normal: 10-55 pg/mL) y fósforo (5,99 mg/dL, valor normal: 2,8-4,5 mg/dL), así como niveles bajos de calcio (7,81 mg/dL, valor normal: 8,5-10,2 mg/dL) y vitamina D (17,6 ng/mL, valor de referencia <20 ng/mL).
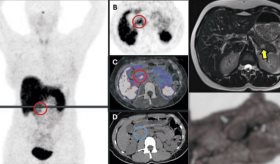
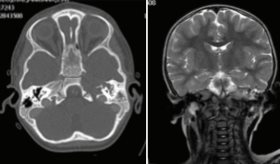
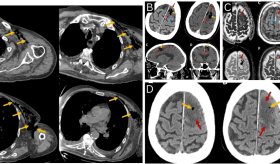
Una tomografía computarizada del cráneo reveló calcificaciones en la sustancia blanca periventricular y en la cisterna perimesencefálica.
En la consulta ambulatoria de endocrinología, el paciente presentó signos físicos evidentes como baja estatura (150 cm) y braquidactilia, con un índice de masa corporal de 26,1 kg/m², lo que indicaba obesidad.
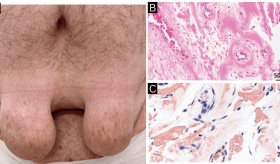
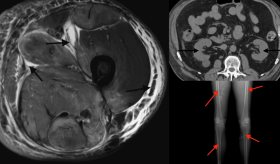
Se realizaron estudios complementarios, incluyendo una densitometría ósea, que mostró una puntuación Z corregida por edad y sexo de -2,1 en la columna lumbar y el cuello del fémur, sugiriendo osteopenia. Las radiografías de la columna vertebral mostraron fracturas vertebrales en L3 y L4, características de fragilidad ósea, además de osteocondromas en el peroné y el antebrazo derecho, indicativos de un trastorno esquelético.
Se solicitó una serie de análisis hormonales, cuyos resultados revelaron niveles elevados de TSH (7,54 UI/mL, valor de referencia: 0,4-4,5 UI/mL), lo que sugería hipotiroidismo subclínico, y niveles normales de T4 libre (0,96 ng/dL, valor de referencia: 0,9-2,3 ng/dL).
También se observaron alteraciones en los niveles de gonadotropinas (FSH 4,23 UI/mL y LH 3,22 UI/mL) y testosterona (5,41 ng/mL), además de microcalcificaciones testiculares, lo que generó sospechas sobre la posible existencia de un trastorno genético subyacente.
Con base en los hallazgos clínicos, radiológicos y hormonales, se sospechó de pseudohipoparatiroidismo tipo 1A (PHP1A), una enfermedad rara que afecta la señalización de la hormona paratiroidea (PTH).
Se realizó un análisis genético, cuyo resultado mostró una variante patogénica heterocigota en el gen GNAS (c165_166insTCAT), que provoca un cambio en la proteína G, esencial para la señalización de la PTH.
Esta variante genética resulta en una forma truncada de la proteína que impide su función adecuada, alterando la regulación de los niveles de calcio y fósforo en el organismo.
El tratamiento consistió en la administración de calcio oral (carbonato cálcico) a dosis de 600 mg cada 4 horas, calcitriol (0,5 mcg cada 8 horas), y hidróxido de aluminio como quelante del fósforo con cada comida.
También se ajustó la dosis de levotiroxina para tratar el hipotiroidismo subclínico, aumentando la dosis total semanal en un 25%. Además, se organizó un seguimiento multidisciplinario con nutrición, nefrología y asesoramiento genético, aunque no se encontraron casos similares en la historia familiar del paciente.
A los 12 meses de tratamiento, el paciente mostró una respuesta terapéutica favorable. Los niveles de PTH disminuyeron a 185 pg/mL, el fósforo se redujo a 5,09 mg/dL, el calcio se normalizó a 9,6 mg/dL, y los niveles de 25-hidroxivitamina D aumentaron a 23,1 ng/mL.
El pseudohipoparatiroidismo (PHP) es una rara enfermedad genética que afecta el metabolismo del calcio y fósforo, caracterizándose por una resistencia a la acción de la hormona paratiroidea (PTH), que regula estos procesos.
Existen diferentes subtipos de PHP, como el PHP1A y el PHP1B, que se relacionan con alteraciones en el gen GNAS, que codifica una subunidad de la proteína Gas, involucrada en diversas vías de señalización hormonal.
Los pacientes con PHP1A suelen presentar resistencia a varias hormonas acopladas a Gas, como la PTH, la TSH, y las hormonas sexuales, lo que lleva a una serie de características clínicas, incluyendo obesidad de inicio temprano, retrasos en el desarrollo y alteraciones cognitivas.
En comparación, el PHP1B está relacionado con defectos en la metilación del locus GNAS y se asocia con una menor cantidad de alteraciones fenotípicas, como la ausencia de obesidad.
En este caso (Cándida Díaz-Brochero, et al) se diagnosticó al paciente con PHP1A debido a una mutación genética en el gen GNAS, que se heredó de forma autosómica dominante.
Además de la resistencia a la PTH, el paciente presentó características como hipotiroidismo secundario a resistencia a la TSH, enanismo y alteraciones en el desarrollo neurológico. La osteoporosis grave del paciente se atribuye a la deficiencia de vitamina D, hipogonadismo leve y resistencia a la hormona de crecimiento.
El diagnóstico de PHP es complejo y requiere un enfoque multidisciplinario, con pruebas genéticas y una revisión detallada de los antecedentes médicos y los hallazgos clínicos.
Aunque se han reportado pocos casos en América Latina, es fundamental aumentar la conciencia sobre esta enfermedad rara para mejorar su diagnóstico y manejo.